We develop novel machine learning graph-theoretic/statistical techniques for solving problems in computational biology, medicine and pharmacology. These techniques can provide an answer to many challenges in these areas, because they can discover patterns and regularities in large amount of noisy data and integrate information from different sources.
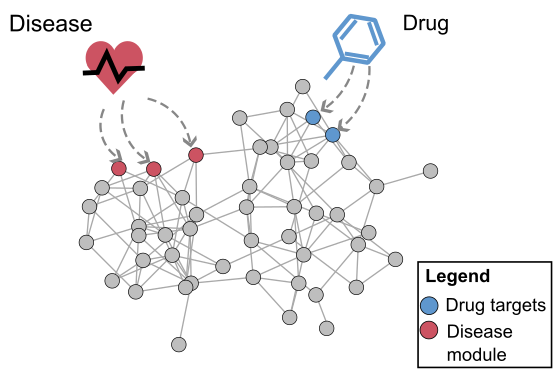
An important idea that has emerged recently is that a cell can be viewed as a complex network of inter-relating proteins, nucleic acids and other bio-molecules. A bio-molecular network can be viewed as a collection of nodes, representing the bio-molecules, connected by links, representing relations between the bio-molecules. Examples of biological networks are, for example, regulatory networks, metabolic networks and signalling networks. At the same time, data generated by large-scale experiments often have a natural representation as networks such as protein-protein interaction (PPI) networks, genetic interaction networks, and co-expression networks. Finally, it is understood that the interconnectivity between cellular components (genes, metabolites, microRNAs etc) has important implications for diseases. The view that has become widely accepted is that genetic disease is the result of abnormal interactions between multiple players in complex networks. From a computational point of view, a central objective for systems biology, medicine and pharmacology is therefore to develop methods for inferring networks or relations between networks.
Much of our research focuses on developing novel mathematical methods specifically suited for making inferences on biological networks, building on these most recent results from computer science and machine learning. In particular, the methods we develop take into account both the structure of the networks representing the data and the structure of the network representing the biological question being answered. The final goal is to be able to answer questions in systems biology and medicine that will help us understand and predict complex cellular behaviour in health and disease.